Карбоновые кислоты
Получение карбоновых кислот
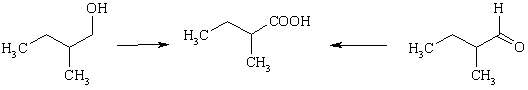
Реакции окисления
Ввиду того, что атом углерода карбоксильной группы находится в максимально возможной для функциональных групп степени окисления, равной +3, кислоты могут быть получены окислением терминального звена углеродной цепи. Действительно, спирты и альдегиды (но не алканы) легко окисляются до карбоновых кислот. Для этого используют широкий спектр окислителей – от таких мягких, как соли CuII до сильнейших (CrO3 в серной кислоте, KMnO4). Как правило, альдегиды окисляются легче спиртов, поэтому окисление первичного спирта сильными окислителями идет до карбоновой кислоты, не останавливаясь на стадии промежуточного образования альдегида.
Кислоты ароматического ряда могут быть получены окислением углеводородных боковых цепей. Это связано с тем, что на первой стадии окисления происходит одного электрона или отрыв гидрид-иона от α-углеродного атома и образование катион-радикала или катиона, соответственно, которые резонансно-стабилизированны ароматическим кольцом. При окислении боковой цепи, содержащей 2, 3 и более атомов углерода, в карбоксил превращается лишь связанный с кольцом С-атом, все другие окислятся до СО2 и Н2О («отгорание боковой цепи»). Среди ароматических соединений легче всего окисляются альдегиды, особенно, орто-замещенные. Бензальдегид быстро превращается в бензойную кислоту на воздухе. см. главу Карбонильные соединения)
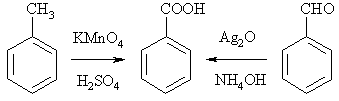
Синтез с помощью металлоорганических соединений
Как известно, металлоорганические соединения, например, реактивы Гриньяра, являются сильными нуклеофилами. Они реагируют даже с таким слабым электрофилом, как CO2. Так, в результате обработки магнийорганического соединения углекислым газом образуется смешанный бромид-карбоксилат магния, из которого карбоновую кислоту выделяют гидролизом в слабо кислой среде (например, в водном растворе хлорида аммония).
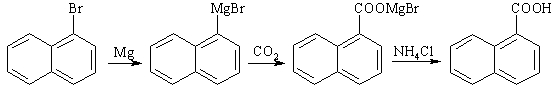
Этот метод синтеза кислот особенно часто применяется в тех случаях, когда бромпроизводное является доступным.
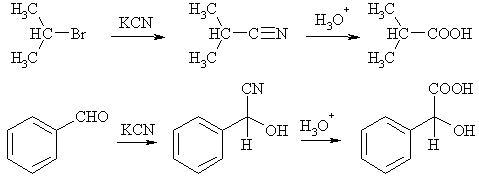
Гидролиз нитрилов и других функциональных производных карбоновых кислот
Практически все соединения, которые объединены в классы производных карбоновых кислот, реагируют с водой в присутствии кислот или щелочей, т.е. подвергаются гидролизу. Часто применяют кислотный гидролиз нитрилов, т.к. последние могут быть получены из других классов органических соединений – галогенидов, альдегидов и кетонов.
Строение карбоксила и карбоксилат-иона
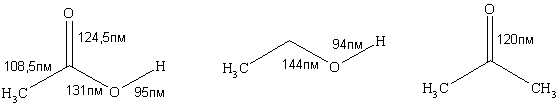
ОН-Кислотность карбоксила намного превышает таковую гидрокисльной группы спиртов и фенолов. Бóлее высокую степень диссоциации карбоновых кислот по сравнению со спиртами объясняют несколькими причинами.
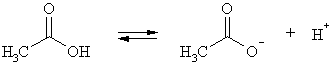
Во-первых, связь O-H в кислотах более поляризована, чем в спиртах. Это обусловлено отрицательным индуктивным и мезомерным эффектом соседней карбонильной группы. Мезомерный эффект заключается в частичном переносе неподеленной электронной пары атома кислорода гидроксильной группы на ее π-связь. Это можно выразить полярной резонансной структурой:
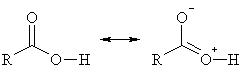
На гидроксильном кислороде наводится частичный положительный заряд, который поляризует связь О-Н и облегчает протонизацию атома водорода.
Во-вторых, как показывают электронографические исследования, оба атома кислорода в карбоксилат-ионе эквивалентны, длина связи С-О имеет величину 0.128 нм. Это свидетельствует о том, что карбоксилат-ион резонансно стабилизирован (в отличие от алкоголят-иона, где отрицательный заряд локализован на одном атоме). Делокализация заряда в значительной степени способствует увеличению кислотности, так как снижает энергию частицы, делая ее более стабильной. А чем выгоднее продукты реакции с точки зрения термодинамики, тем равновесие более смещено вправо.
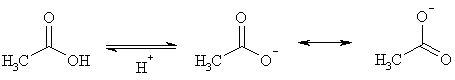
Карбонильный атом кислорода карбоксильной группы, как и в альдегидах, обладает двумя несвязывающими парами электронов и, может координироваться с кислотами Льюиса. Его -I и –М-эффекты наводят на карбоксильном атоме углерода положительный заряд, который, с одной стороны, усиливается –I-эффектом, а, с другой стороны, ослабляется +М-эффектом атома кислорода гидроксильной группы. Это делает плоский карбоксильный атом углерода центром, удобным для атаки нуклеофилами. Однако, влияние мезомерного эффекта группы ОН более ощутимо по сравнению с индуктивным, поэтому электрофильность атома углерода карбоксигруппы намного ниже, чем электрофильность альдегидного (кетонного) карбонила. Предварительное протонирование карбонильного кислорода кислот или координация с кислотами Льюиса повышает на атоме углерода положительный заряд и его электрофильность резко усиливаются.
Карбоксильная группа является акцептором электронов (-I-эффект) по отношению к α-углеродному атому и протонам, связанным с ним. Это делает возможным отщепление α-протона и его миграцию на атом кислорода, т.е. явление кето-енольной таутомерии.
Перечисленное выше объясняет разнообразие химических свойст карбоновых кислот: сравнительно высокие температуры кипения, растворимость в воде низших гомологов, заметную ОН-кислотность, способность к восприятию электрофильной атаки карбонильным атомом кислорода (основность), к нуклеофильному замещению гидроксила, превращениям по α-углеродному атому.
|
|
||
|
|
|
|